

CASE OF THE MONTH: June 2023
Progressive Vision Loss in a Patient with Ataxia



A 55-year-old male with history of hypertension and coronary artery disease presented to the clinic complaining of blurred central vision in both eyes that had slowly progressed over the last 10 years. The patient additionally noted progressive lower extremity weakness and gait instability that began 5 years prior. His best corrected visual acuity was 20/125 in each eye. Intraocular pressure was normal in each eye. There was no afferent pupillary defect. Anterior segment examination was unremarkable. Fundus examination showed pigment epithelial mottling in the central macula of each eye, and optical coherence tomography (OCT) revealed focal subfoveal disruption of outer retinal layers in both eyes (Figure 1). Magnetic resonance imaging (MRI) demonstrated cerebellar atrophy along with microvascular ischemic changes (Figure 2).
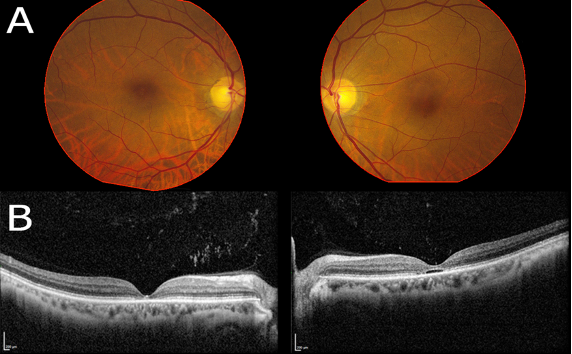
Figure 1. Color fundus photographs of the right and left eye (A) demonstrating subtle macular pigmentary changes. B. Optical coherence tomography of the right and left eye demonstrating focal subfoveal loss of the ellipsoid zone with outer retinal atrophy in both eyes and cavitation in the left eye.
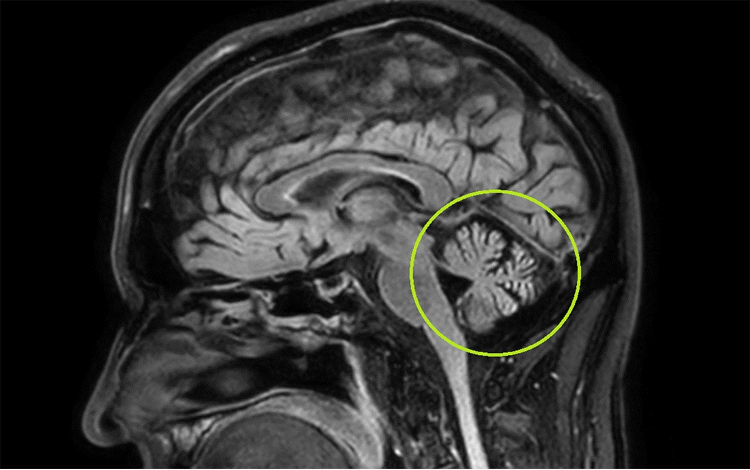
Figure 2. Sagittal T1 magnetic resonance imaging (MRI) of the brain demonstrating cerebellar atrophy (green circle).
Genetic testing revealed 44 CAG repeats in the ataxin 1 (ATXN1) gene, consistent with a diagnosis of spinocerebellar ataxia type 1 (SCA-1).
Spinocerebellar ataxia (SCA) is a group of rare neurodegenerative disorders characterized by oculomotor disturbances, ataxia, and dysarthria1. SCA Type 1 is the result of CAG trinucleotide expansion in the ATXN1 gene, leading to accumulation of cytotoxic polyglutamine products1. Both the severity and age of disease onset are directly correlated to the number of repeats, with expansion of repeats resulting in anticipation with younger age of symptom onset in successive generations2.
While SCA type 7 has been associated with cone dystrophy and is the form of SCA classically linked with retinal involvement3, SCA type 1 conversely has been rarely implicated in ophthalmic disease. One report of 6 patients in 3 families showed progressive visual deterioration with optic atrophy and decreased corneal endothelial cell density4. An additional case report noted reduction in both rod and cone responses on ERG suggestive of a cone-rod dystrophy5.
More recently, studies have described subtle macular changes best appreciated on OCT. A series described four patients in a single family with altered foveal lamination and subfoveal focal disruption of outer retinal layers similar to our patient6. A additional report described 2 patients with SCA-1 with similar subfoveal hyperreflective outer retinal cavities with global thinning of the underlying neuroretina that were associated with central scotoma and depression of central electrical potentials on multifocal electroretinogram7. Additional recent series demonstrated similar focal ellipsoid zone loss in 5 patients8, and an additional report noted similar findings in 4 out of 5 patients with confirmed SCA-19. A case study noted progression of the area of ellipsoid zone loss over a period of 30 months that worsened more quickly than other macular degenerative diseases10.
1. Pula JH, Gomez CM and Kattah JC. Ophthalmologic features of the common spinocerebellar ataxias. Curr Opin Ophthalmol 2010; 21:447-453.
2. Stevanin G, Dürr A and Brice A. Clinical and molecular advances in autosomal dominant cerebellar ataxias: from genotype to phenotype and physiopathology. Eur J Hum Genet 2000; 8:4-18.
3. Hugosson T, Gränse L, Ponjavic V and Andréasson S. Macular dysfunction and morphology in spinocerebellar ataxia type 7 (SCA 7). Ophthalmic Genet 2009; 30:1-6.
4. Abe T, Abe K, Aoki M et al. Ocular changes in patients with spinocerebellar degeneration and repeated trinucleotide expansion of spinocerebellar ataxia type 1 gene. Arch Ophthalmol 1997; 115:231-236.
5. Thurtell MJ, Biousse V and Newman NJ. Rod-cone dystrophy in spinocerebellar ataxia type 1. Arch Ophthalmol 2011; 129:956-958.
6. Lebranchu P, Le Meur G, Magot A et al. Maculopathy and spinocerebellar ataxia type 1: a new association? J Neuroophthalmol 2013; 33:225-231.
7. Vaclavik V, Borruat FX, Ambresin A and Munier FL. Novel maculopathy in patients with spinocerebellar ataxia type 1 autofluorescence findings and functional characteristics. JAMA Ophthalmol 2013; 131:536-538.
8. Oertel FC, Zeitz O, Rönnefarth M et al. Functionally Relevant Maculopathy and Optic Atrophy in Spinocerebellar Ataxia Type 1. Mov Disord Clin Pract 2020; 7:502-508.
9. Nishiguchi KM, Aoki M, Nakazawa T and Abe T. Macular degeneration as a common cause of visual loss in spinocerebellar ataxia type 1 (SCA1) patients. Ophthalmic Genet 2019; 40:49-53.
10. Hirose A, Katagiri S, Hayashi T et al. Progress of macular atrophy during 30 months' follow-up in a patient with spinocerebellar ataxia type1 (SCA1). Doc Ophthalmol 2020.